Electronic bands – the energy solutions εi(k) of the Kohn-Sham equation of a non-interacting electron in an effective potential, are, despite of approximations made in practical DFT calculations, very successful in describing the physics of condensed matter. It is sometimes necessary (or simply practical) to describe the band-structure (“Spaghetti”) in terms of local orbitals, that is in a language that is more close to the “chemist’s picture” of atomic s,p,d orbitals. Generally, the s,p,d nomenclature is linked to the angular momentum l of a radially symmetrical potential (“Hydrogen model”) – and not a periodic crystal potential. It can be shown, however, that the resulting electronic bands can often be assigned to a certain l, at least for the low-energy core bands.
Most programs available allow to obtain this information either as a function of energy alone (the density of states, DOS and projected density of states, PDOS) or resolved in energy and crystal momentum which is often called a “fat-band” representation as the weight of the orbital s,p,d characteristic is shown as the line thickness for practical reasons. As a simple example I have calculate the PDOS of bulk silver using the ELK code, a full-potential (all-electrons) linearised augmented-plane wave (FP-LAPW) DFT code. The relevant task numbers in this case are 0 (the ground-state), 10 (the total and projected density of states) and 21 which the manual lists conveniently to give us the Band structure plot which includes angular momentum characters for every atom. Details can be found in the attached elk.in file.

The figure shows the band structure on the left-hand side and the corresponding integrated information with the same color-code on the right-hand side. I included arrows to highlight how certain features in the PDOS can be interpreted based on the band structure and vice versa. The first thing to notice is that most bands above the Fermi level (set as 0 in the ELK code) are a mix of various l-states whereas just below the Fermi level the d-states form a fairly flat (localized) set of bands.
Now I usually work with the plane-wave pseudopotential method as implemented in Quantum-Espresso (QE 6.3) and not with full-potentials but I chose above calculation as an entry point to show what can be a problem with pseudopotentials – bear with me. Turning to QE I generate the same input structure as previously used in ELK using cif2cell with the program switch -p pwscf. I use my favorite set of norm-conserving pseudopotentials from the ONCV library and generate the wave-functions using the latest version (3.3.1) of the ONCV code. For silver I find that a regular k-point grid of 11x11x11 and a cutoff of the wave-functions of about 80 Ry gives converged results. The calculation of fatbands is a bit more difficult in QE than ELK and involves the following steps:
- calculation of the band-structure using pw.x and bands.x
- calculation of the “projection on wave-functions” using projwfc.x
- generation of the weights by calculating the overlap between projections and wave functions using plotband.x
All steps are automated in the bands.sh script attached but I want to highlight that the setting of lsym=.true./.false. in the input for bands.x and projwfc.x is important. When the projection file is found by plotband.x it asks for a list of wave functions to calculate the overlaps. The proper numbers can be found in the output of projwfc.x and looks something like
state # 1: atom 1 (Ag ), wfc 1 (l=0 m= 1)
state # 2: atom 1 (Ag ), wfc 2 (l=1 m= 1)
state # 3: atom 1 (Ag ), wfc 2 (l=1 m= 2)
state # 4: atom 1 (Ag ), wfc 2 (l=1 m= 3)
state # 5: atom 1 (Ag ), wfc 3 (l=0 m= 1)
state # 6: atom 1 (Ag ), wfc 4 (l=2 m= 1)
state # 7: atom 1 (Ag ), wfc 4 (l=2 m= 2)
state # 8: atom 1 (Ag ), wfc 4 (l=2 m= 3)
state # 9: atom 1 (Ag ), wfc 4 (l=2 m= 4)
state # 10: atom 1 (Ag ), wfc 4 (l=2 m= 5)
The ordering here is the same as in the pseudopotential file, so in case there is any doubt consult the header of the pseudo and confirm the valence configuration:
#n l occ
4 0 2.00
4 1 6.00
4 2 10.00
5 0 1.00
The following figure shows the fatbands as obtained by QE and the band structure obtained from maximally localized wannier functions (MLWF) via wannier90.
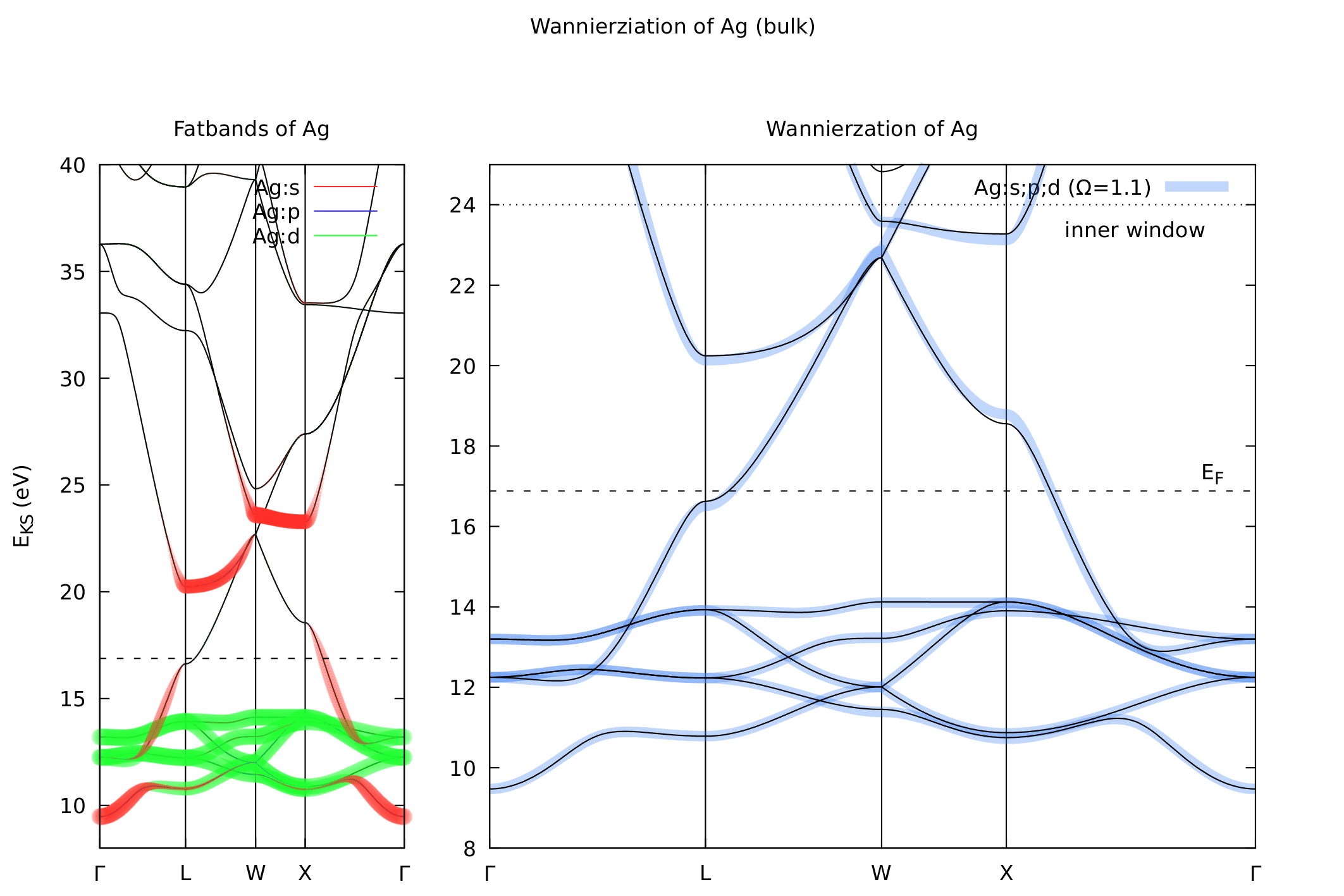
If you are familiar with wannier90 you know that the definition of an initial “guess” of wavefunctions is usually critical in order to obtained well localized wannier functions. Looking at the fatbands of QE the character of the bands just above the Fermi level remains, unfortunately a mystery. Overall the band-structure agrees very well with the results obtained from ELK which tells us that the pseudo has no problem but the wave functions generated with ONCV are less than perfect. If I had read the release notes carefully I would have been able to expect this:
The upf output now includes pseudo wave funtions for the occupied states.
This will allow LDA+U calculations in some codes. Please note that
ONCVPSP potentials have not been tested or benchmarked for this use.
Also be aware that LDA+U is a semi-empirical mean field theory, and
does NOT represent many-body physics.
Ultimately I had no problem defining proper projections (with the guidance of the ELK fatbands) but above should be kept in mind when performing, for example, LDA+U calculations in QE (note that this does not really pose a problem here as the highest-l manifold, the d-bands, have proper wave-functions!).
For wannier90 initial projections of
begin projections
Ag: s;p;d
end projections
result in an average spread of Ω~1.1 per atom which is fairly well localized and the resulting tight-binding bands agree well with the DFT bands, as can be seen on the right-hand side above.
Hope this was helpful – if there is any problem running attached scripts please leave it in the comments!
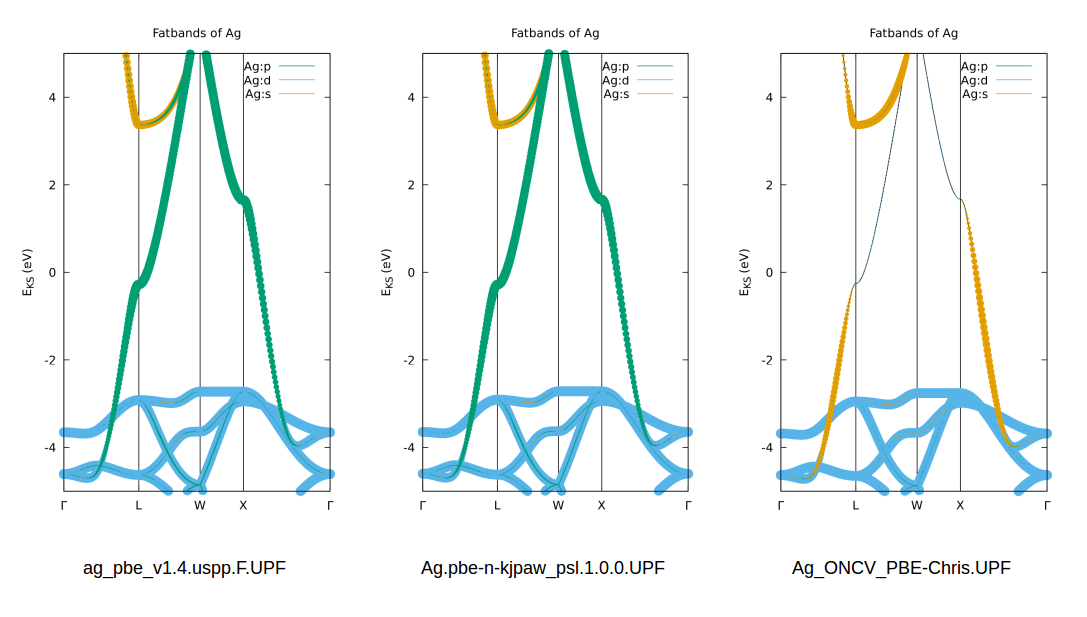
PS: as correctly mentioned in the comments, a “better” pseudopotential would be able to produce a more accurate fat-band representation. Therefore I attached a small comparison of 3 easily available PPs (from the PSLibrary, the GBRV library and ONCV library) – an updated “run script” is attached as well. HTH!

7 responses to “From “Fatbands” to Wannier Functions of Silver”
My 2cents of comments are not coming from an expert, but I will try to say something that might be useful. From your .zip file, which is not included the actual pseudopotential you generated namely: Ag_ONCV_PBE-Chris.UPF, this is important to see how it works in projected band plot with pwscf. The p orbital characters are missing in the fatband plot may be coming from the generated pseudopotential, result can be improved if we use QE’s pseudopotential, for examples: https://www.quantum-espresso.org/upf_files/Ag.pbe-dn-rrkjus_psl.0.1.UPF
And it’s even more helpful if the provided zip file contains also the “plot.gnuscf” as stated in the bash file.
Cheers 🙂
Delong.
LikeLiked by 1 person
Hi Delong, thank you for your comment and you are absolutely right. I found that the PSLibrary (1.0.0 is the most recent version I believe) has much better wavefunctions than the ONCV and the GBRV pseudos are also giving much improved “fatbands”.
The reason I am usually sticking with the ONCV is that, in my experience, they give good structural (lattice constants, phonons) results and post-DFT codes such as YAMBO require norm-conserving pseudos if I remember correctly. So there is a certain “pitfall” when blindly trusting a single set of pseudos to satisfy “all needs” 🙂
Thanks for your comment, I should probably give a comparison of fatbands somewhere!
LikeLike
Agree with the second comment.
LikeLike
Dear Christoph,
How could one include spin-orbit couplings with fat band?
Thanks!
SD
LikeLike
Many thanks. it’s even more helpful if you also attach the “plot.gnuscf” file as stated in the bash file
LikeLike
Hi Cristoph,
How do you define the projections for wannier input file? I have not found a comprehensive way or easy explanation on how to define them for Ca, Sr, Ba and B. Please help. I am working with hexaborides (CaB6, SrB6, BaB6) and I am using your proposed workflow for calculating band structures in Wannier90, since I am dealing with hybrid functionals. For some reason that it is unknown to me the autoprojections is not working, hence I need to the define the projections manually.
Thank you,
LikeLike
Hello there.
I may be going off topic by asking this but since I am a newbie in this field I hope you will not mind me asking this. I want to know how you actually plotted the data to get those graphs. Actually I am also working with both ELK and Qunatum-Espresso and I have plotted fatbands from the output of ELK using Origin-Pro but I don’t think that is what you used to get those beautiful graphs.
So could you tell me which program you used and if possible a learning guide/reference material as well.!
LikeLike