In this short tutorial I will show how to first perform a QE calculation of the molecule benzene in a vacuum box at the GGA level. Then, we will use YAMBO to calculate a single shot G0W0 correction to the Kohn-Sham eigenvalues. As a last step we calculate the density of states at the GW level as well as a projected density of states on the C:s orbitals.
The main purpose for this tutorial is to demonstrate the work-flow based on instructions by Daniele Varsano (here).
I will assume, for the sake of keeping this tutorial short, that one already knows the basic steps of creating the base folder for a YAMBO calculation by first performing an SCF and NSCF calculation. The crucial step for the GW-PDOS calculation is to perform a PDOS calculation using projwfc.x before creating the YAMBO base folder in p2y. That means the proper sequence is: scf (pw.x) > nscf (pw.x) >pdos (projwfc.x) > p2y > yambo initialization > yambo G0W0> ypp. After the p2y step there should be a new file in the base folder called ns.atom_proj_pwscf – this basically indicates that p2y found atom projections and created a database.
As always: this is a basic example and not a converged calculation (this will become obvious right away)
Let’s first see what the G0W0 (yambo -x -p p -g n) calculation does to the eigenvalues:
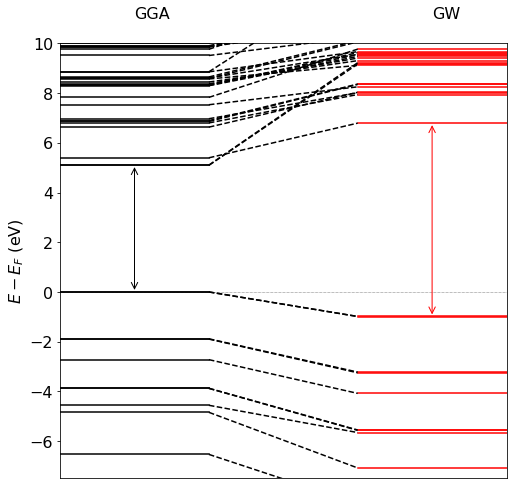
As you can see in above plot of the o.qp file, the gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied MO (LUMO) at the GGA level is around 5 eV. This increases to around 8 eV at the G0W0 level (shown in red). A converged GW gap for this molecule is around 10 eV. [1]
Ignoring this discrepancy for now we move to the generation of a DOS input using ypp -s s -V all, increasing the verbosity to all helps with defining the input. A few lines deserve special attention:
To tell ypp that we attempt to create atom projected DOS we change the project mode line to: PROJECT_mode= “ATOM”. By itself, the GW corrected database will not be read, therefore the line related to that reads: GfnQPdb= “none”
last but not least you will see a larger block at the bottom of the file related to selecting atomic wave functions – I will come to that in a second.
To find out which atom types and wave functions are in our calculation we can take a look at the report generated during the initialization of the calculation:
[02.01] Unit cells
==================
Cell kind : CUB
Atoms in the cell : C H
number of C atoms : 6
number of H atoms : 6
List of atomic masses
Mass of C : 12.01070
Mass of H : 1.000000As we can see, there are two types (C, H) and 6 atoms each (C6H6). The H pseudo potential will contain the 1s wave function with angular quantum number l=0, C will contain the 2s (l=0) and 2p (l=1). If we don’t care about the type of atom and wave function we can just leave them set to -1 and run ypp. This will generate the total DOS at the GGA level.
Advancing one step, we can insert the QP corrections by replacing GfnQPdb= “none” with
GfnQPdb= "E < ./SAVE/ndb.QP"Note that the ndb.QP can also lie in a different folder if the GW step was called using a job identifier (-J)! The default folder is however SAVE. Running ypp one more time will now tell us in the output that it will apply corrections to the eigenvalues:
[QP_apply] Action to be applied: E<./G0W0/ndb.QP[ PPA@E 27.21138 * XG 949 * Xb 1-100 * Scb 1-100]The last but very interesting step is to not calculate only the total DOS but select a specific atom type and angular character, for example the H:s or C:s or C:p. This can be done by changing the part at the bottom of the ypp input file:
% PDOS_kinds
1 |1 |Allows us to project onto the first type of atom, which is carbon. The next setting
% PDOS_l
0 |0 |Allows us to project onto the s (l=0) orbital of that atom. More fine-grained selections can be defined. The resulting data can be summarized in the following DOS plot
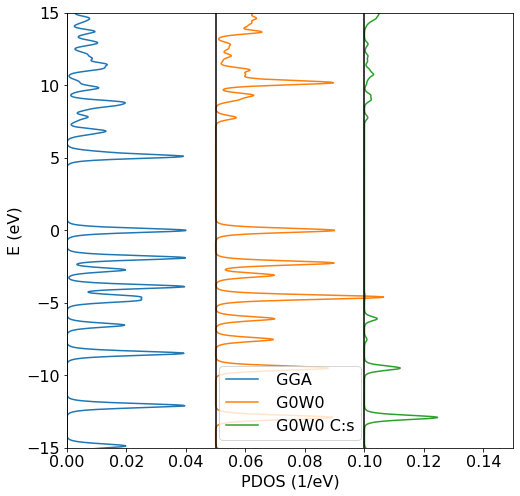
Shown are the GGA DOS (blue), which exhibits a significantly smaller gap than expected, the G0W0 DOS in orange as well as the C:s projected G0W0 DOS in green. Note that the DOS is aligned so that 0 eV corresponds to the highest occupied orbital.
A script to run the calculations and produce the plot (using gnuplot) is attached – have fun reproducing the results yourself!
[1] Neaton et al., https://arxiv.org/abs/cond-mat/0606640

One response to “GW DOS calculations using QE and YAMBO”
Thanks for information Kehidupan Kampus
LikeLike